Documentation
This step-by-step tutorial will let IsoQuaC analyze your labeled LC-MS/MS proteomics differential expression experiment and provide you with output files on both the non-redundant-peptide level as well as protein level, including a quality control and differential expression report in PDF format.
-
You'll need the peptide-spectrum matches in .txt, .csv/tsv or .xlsx format. These should be in separate files: one per LC-MS/MS run. Please verify that all required data 'observables' or 'columns' are present in your PSM files and have valid contents, especially if you're not using files generated by Proteome Discoverer (PD).
- Sequence: {string} the peptide sequence
- Master Protein Accessions: {string;string;...} accession numbers of the proteins associated with a PSM. separated by semi-colons.
- First Scan: {integer} identifier for the MS scan in which the peptide was detected. If you don't have such a column, just create one with all values set to a unique integer. PSMs detected by multiple times by different PSM Algorithms should have the same value.
- (???): {string} names of the reporter labels or channel names you provided in the DoE file.
If this is not the case, simply create a text file (wrapper) with on each line YOUR_NAME for the observable, as well as OUR_NAME, separated by a TAB
If you have any additional columns or observables you would like IsoQuaC to use, please refer to the complete set of compatible column names.
-
You'll also need a 'design of experiment' (DoE) file. Create one by going to the "New DoE" page and following the instructions. You'll have to give names to your different 'LC-MS/MS runs', specify which biological/chemical conditions were used in which MS run, and indicate for each MS run which 'channels' or 'reporter labels' (as they appear in the headers of your PSM files) belong to which of those conditions. At the end of the procedure, you can download the DoE file you need. This file can easily be modified with a text editor if you want to correct a mistake, or quickly generate your own new DoE file.
-
Got your PSM files and DoE file? Great, in the "New Job" form, choose a job name. Then, upload your DoE file.
-
Fill out the Job Settings form. Please remark:
- If you are not using Proteome Discoverer for generating PSM files, you will be warned to provide the wrapper file(s) you (should have) created in the PSM file verification step by uploading one under the "Advanced" button for each PSM data file.
- In case you used multiple search engines to generate PSMs, the information from the Master engine will be preferred over that of the Slave(s).
- IsoQuaC will refrain from using 'shared peptides' (peptides that may correspond to multiple proteins) unless you check the box under "Advanced settings". In that case a second analysis will be done for the assuming that all proteins make (full) use of every peptide they share with another protein.
-
Press "Submit job" and make sure you land on the "Job Info" page which states your job is running. If you didn't provide an e-mail address, be sure to copy and save the job ID. You can use your job ID to retrieve your job information from the "Find job" page in case you close your Job Info page.
-
As soon as the job is done, the Job Info page will provide you with download links for the report file as well a .zip file containing:
- normalized data on the non-redundant, modified peptide level (1 file per LC-MS/MS run)
- differential expression analysis on the protein level (in 1 comprehensive file as well as split out in 1 file per condition)
Here you can find the scientific description as well as prerequisites and technical specifications of the IsoQuaC workflow, which runs on a server located at UHasselt and is accessible through a web interface.
The scientific workflow consists of three distinct steps:
- Processing: cleans the input data while gathering some QC information, and aggregates the PSMs to the modified, non-redundant peptide level, then performs normalization.
- Analysis: while gathering additional QC information, transforms the information from the peptide to the protein level and performs a differential expression analysis (DEA). Also performs an exploratory analysis for quality control (QC) purposes, including a principal component analysis (PCA) and hierarchical clustering (HC) on the peptide-level data matrix.
- Reporting: produces a PCA plot (first 2 components), HC dendrogram and for each non-reference condition a volcano plot with a list of the top differential proteins. From the gathered QC info, produces some statistics and MS1 calibration and intensity plots if possible. Lastly, Summarizes all visualizations, statistics and other relevant information (including meta-data) into a PDF report.
The output for the user contains the created report, as well as some tab-separated (.tsv) data files:
- normalized data on the non-redundant, modified peptide level (1 file per LC-MS/MS run)
- differential expression analysis on the protein level (in 1 comprehensive file as well as split out in 1 file per condition)
If you wish to acquire any intermediate output files, please contact us.
Most of the parameters which control the behaviour of the workflow are fixed when using the web interface. If you wish to run a job with a custom set of parameters, please contact us.
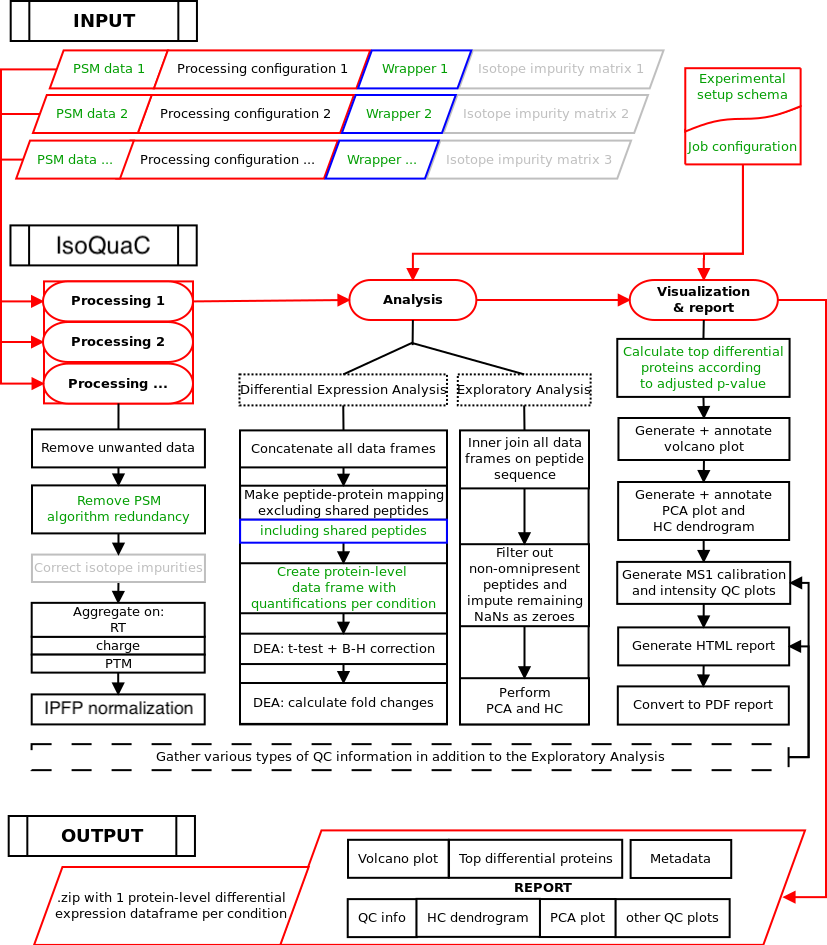
IsoQuaC workflow overview. Red: main input/output and workflow backbone. Blue: optional input/output and workflow steps. Green: input and workflow steps directly influenced by user-specifiable parameters. Grey: currently disabled.
Prerequisites
The following requirements are to be met in order to use IsoQuaC:
- This workflow was built for labeled shotgun proteomics with TMT-labeled N-plex peptide samples eluted through a liquid chromatography column, then ionised, then selected for m/z value by an MS1 and then further fragmented to be quantified by an MS2. However, it also works for experiments based on equivalent techniques, f.i. iTRAQ labeling, and it has been shown (publication pending) to also work for Next-Gen Sequencing (NGS) count data, f.i. raw RNA-Seq counts.
- The MS2 spectra of the mass spectrometer should have already been analyzed and converted into peptide-spectrum matches (PSMs), for instance using one or multiple database search algorithms like, for instance, SEQUEST [DOI: 10.1016/1044-0305(94)80016-2] or MASCOT [DOI: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2].
- In case multiple samples belonging to the same condition are spread across
multiple LC-MS/MS runs,
make sure that the samples belonging to the same condition are always evenly spread
across all the LC-MS/MS runs they appear in.
Do not, f.i., put 5 samples of condition A and 1 of condition B in the first LC-MS/MS run, and then 1 sample of condition A and 5 of condition B in the second. Rather, you should respect intelligent experimental design and spread the samples so that each run contains 3 samples of each condition. - IsoQuaC normalises the data through an iterative proportional fitting procedure (IPFP). The normalisation can be verified on an MA-plot of your peptide- or protein-level data:
- The majority of proteins/peptides are not differentially expressed,
to avoid a bias in the estimate of the mean value. It is assumed that up-down shifts
are not due to biological causes. The reference set used in the normalization step
is the set of all peptides identified in the experiment.
MA-plot: the observations form a single 'cloud' with a dense center and less dense edges, as opposed to, for instance, two clouds or a cloud with uniform density. - The number of up-regulated proteins/peptides is roughly equal
to the number of down-regulated ones. If the data were skewed, this
would lead to a bias in the normalization result.
MA-plot: the cloud of observations exhibits a bilateral symmetry about some axis (usually horizontal, but not necessarily). - The systematic bias in each sample's quantification signal
intensity is linearly proportional to the magnitude of the intensity.
Only this way it is possible to find one appropriate normalization factor for each
quantification sample.
MA-plot: the axis of bilateral symmetry is a straight line (which may be inclined), i.e., the moving average M-values of the central cloud form a straight line.
- The majority of proteins/peptides are not differentially expressed,
to avoid a bias in the estimate of the mean value. It is assumed that up-down shifts
are not due to biological causes. The reference set used in the normalization step
is the set of all peptides identified in the experiment.
- We recommend using non-transformed intensities as input values.
Processing
The input data files are PSM files, preferrably exported from Proteome Discoverer (PD) 2.1, although it is perfectly possible to use different platforms as long as the data complies to the specifications. They can be represented as tables (one for each LC-MS/MS run) where each row represents a unique PSM corresponding to a peptide observation, containing the observed values of the variables/quantities described by the column headers.
The following steps process
the data into a human-readabe format at the peptide level that is ready for interpretation by
the data analysis part of the
workflow. Relevant parameters from the processing configuration file (automatically generated by
the web interface) are in teletype font.
Column header or variable names are italicized.
- Read the PSM data file into a Pandas data frame, apply the wrapper and load the processing configuration parameters.
- Remove obsolete (non-wanted) columns/variables.
wantedColumns. - Information about regular Protein Accessions and their corresponding information in the Protein Descriptions strings are removed; only the information about master proteins is kept.
- Remove PSMs that have missing values in
noMissingValuesColumns, or that have missing values in all of thequanColumns. - Remove PSMs with Confidence levels worse than
removeBadConfidence_minimum.removeBadConfidence_bool. - Remove PSMs with Isolation Interference [%] levels higher than
removeIsolationInterference_threshold.removeIsolationInterference_bool. - Clean PTM information by keeping only the PTM identities, not their locations, and removing all 'TMT' label modifications. The remaining PTMs are then sorted alphabetically.
- Remove redundancy due to the use of multiple
PSMEnginePriorityPSM detection algorithms, which are specifiable by the user.removePSMEngineRedundancy_bool. If a peptide observation yielded more than one PSM (one for each algorithm) then we only keep the PSM of the PSM engine with the highest priority, and the others are removed. - (Currently unavailable) Correct isotopic impurities, see Isotope impurity correction.
- Aggregate on retention time RT [min] (RT), see Aggregating duplicate observations.
aggregateRT_bool,aggregate_method. - Aggregate on Charge if possible, see Aggregating duplicate observations.
aggregateCharge_bool,aggregate_method. - Aggregate on post-translational Modifications (PTMs) (Disabled by
default) if possible, see Aggregating duplicate observations.
aggregatePTM_bool,aggregate_method. - Perform the IPFP normalization, see normalization.
- Save all results to disk and pass them on to the analysis step.
For all removed data, at least the columns specified by
removalColumnsToSave are
saved – sometimes together with more information relevant at a certain removal step
– into either one file for all removal steps or into separate files per removal step, as
specified by removedDataInOneFile.
Aggregating duplicate observations
Removing redundancy in the list of PSMs due is what we call aggregating. This is necessary to avoid artificial enrichment by counting the same peptide multiple times in the DEA. We consider three possible variables that cause redundancy, which we may aggregate on:
- RT (
aggregateRT_bool): depending on the data-dependent acquisition (DDA) settings of the MS, it forgets the peptides previously detected by the MS1 after a fixed time interval tDDA (i.e. after each MS1 sweep) and possibly detects the same peptide multiple times at different RT values, if it takes longer than tDDA to elute from the LC column. - Charge (
aggregateCharge_bool): the electrospray ionization is a stochastic process which may place zero or more than one positive charge on each peptide. Since the MS cannot distinguish peptides but only m/z values, the same peptide can be detected multiple times (with different charge values) even within the same MS1 sweep. - PTM (
aggregatePTM_bool): just as charge influences the m/z value, so does the mass of the peptide being detected by the MS1. As a peptide may carry modifications – either multiple TMT labels or biologically relevant modifications like phosphorylation – it may thus appear with different mass values and be detected multiple times even within the same MS1 sweep. Please note that on the peptide level we use the PTM information as-is. Since PD includes PTM location information, this means that the same ligand in a different location is considered a different PTM. On the protein level (later in the workflow), the N-terminal TMT label modifications as well as any PTM location information are filtered out.
The aggregations on charge and/or PTM are optional in theory, but are specified
by their
respective boolean parameters. By default Charge aggregation is enabled (unless the
corresponding column
is missing from the data frame) and PTM aggregation is disabled. The aggregation on RT is
mandatory, as we assume its
variability is never of biological importance. It is always executed, even if the 'RT
[min]'
column does not exist as we assume RT to be the only other source of redundancy.
Therefore, there should be no additional properties causing peptide sequence redundancy in
your data set, except for the use of multiple PSMEnginePriority.
For each aggregation a representative PSM is chosen. This representative is
added to
the data frame and the representees are removed. It contains all the information of the
representee that had the best PSM score, except for the quantification values. The best
PSM score is the best score calculated by the Master PSM algorithm, if available, or
otherwise the best score calculated by (one of) the Slave algorithm(s), if available, or
if no score is available the program just takes the information of the first representee
encountered. The quantification values of the representative can be determined in one
of three ways, specified by aggregate_method:
- {bestMatch}: the quantification values of the PSM with the best PSM score (see above). In case all PSMs have missing scores, the most intense is chosen instead (see below).
- {mostIntense}: the quantification values of the PSM with the highest sum of its MS2 quantification values.
- {mean}: the value for each quantification sample is the mean of the quantification values of all representees for that same quantification sample.
- {geometricMedian}: the components of the quantification vector that is the geometric mean of the quantification vectors of the representees, where the quantification vectors are vectors in Euclidian N-space with their ℓ1-norm set to 1 and with the (re-scaled) quantification values as its components. To calculate the geometric median, we use the algorithm developed by Vardi et al. (DOI: 10.1073/pnas.97.4.1423).
Note that the only information carried by a representative that is (currently) also used in the downstream analysis, is the information identical for the corresponding representees (like the peptide sequence) – except for the quantification values about which we have just elaborated.
Any data that has been removed at any point can be saved to a 'removed data collection' which may be used by the IsoQuaC team only anonymously and only for meta-analysis purposes.
(disabled) Isotope impurity correction
This functionality is currently disabled. Provided with the isotope impurity matrix (not the TMT-style format), one can easily calculate the true quantification values from the observed ones by just solving the linear system of equations as explained by Shadforth et al. (DOI: 10.1186/1471-2164-6-145). Of course this requires one to know the observed quantification values for all of the PSM peaks that receive a contribution from reporter isotopes, and those may not always appear at the same m/z-values as the other labels and hence will not be available in the PD2.1 output (if PD2.1 did not apply the correction itself).
IPFP Normalization
The normalisation is based on an iterative proportional fitting procedure (IPFP), where the mean-values of the rows and the columns of the data matrix are fitted to the value of one. IPFP is also commonly referred to as the RAS-procedure or raking-procedure. Through normalisation, systematic biases in the data are corrected for all peptides across a sample. The algorithms makes two assumptions:
- The relative nature of the quantification values
- The equimolarity of the samples
If P if the number of peptides and N the number of samples, we want to impose two constraints to transform the unnormalized P × N matrix A that contains the quantification values, into a normalized matrix K that does obey these constraints. The first constraint is that the values of each row must sum to N (relative quantification) and on the other hand that the values of each column must sum to P (equimolarity).
To reach convergence for K, we could infinitely iterate. Ofcourse, this is not feasible and therefore one can never guarantee K to be obtained exactly since we are restricted to our assumptions. IPFP has been shown to have a monotonically converging entropy, ℓ1-measure and likelihood. We define the precision after iteration i as N/2 × |Ri−1 − IP |; the number of columns divided by two, times the total deviation of the matrix with row means from the P × P identity matrix. Note that after each iteration, the deviation of the column means is of course exactly zero. The convergence of this algorithm – and thus also the precision improvement – is exponential in the number of iterations, and in fact the convergence is asymptotically identical.
If there happen to be missing values in the quantification matrix A, we just calculate
the numpy.nanmean instead of the mean. This function from the Python package numpy
ignores any missing values in the calculation of the mean, so a row with one missing
value now has a sum of (N − 1) . However, the mean value for the normalized
quantification values in that row will still be 1 and the algorithm needs no further
adaptations.
Analysis
After the processing steps have been performed for each MS run individually, the quantification values are normalized and the data is ready for analysis.
During the analysis steps some metadata is also extracted, some of which will be shown in the report file and some of which is kept by the IsoQuaC team but may only be used anonymously and only for meta-analysis purposes.
The necessary ingredients to make visualizations of the DEA and Quality Control
and a report in general are calculated and extracted in the following steps in the two sections
below. Relevant parameters from the processing configuration file (automatically generated by
the web interface) are in teletype font.
Column header or variable names are italicized.
Differential Expression
- Combine the data frames of all MS runs into one data frame by performing an 'outer join': simply a vertical concatenation but with the quantification columns of each MS run kept separate, still retaining their distinct names. In this way, peptides with the same Sequence can appear multiple times (will be considered a repeated measurement), but no peptide of one MS run has any values in the quantification columns corresponding to other MS runs (those values are 'missing').
- Use latter combined data frame to compile a mapping – across all MS runs –
between the
proteins
found and their respective peptides. This can be done in two ways because some peptides
may possibly correspond to more than one (master) protein, even though the proteins have
already been grouped:
- Each protein gets contributions only from non-shared peptides: they
are uniquely associated to that protein.
minExpression_bool - Each protein gets the full contribution from all peptides that are
associated with it, even if they are in reality shared with other proteins.
fullExpression_bool
- Each protein gets contributions only from non-shared peptides: they
are uniquely associated to that protein.
- Execute the peptide-protein mappings according to previously mentioned dictionary, to obtain a data frame on the protein level. For each protein entry the data frame contains the protein description, the list of (representative) peptides and the list of corresponding quantification values, split into one column for each condition.
- Perform a DE test for each protein using a moderated t-test with Benjamini-Hochberg correction. The p-values and Benjamini-Hochberg adjusted p-values are added to the protein-level data frame.
- Calculate for each protein and each condition the log2 fold
change w.r.t. the
reference condition
referenceConditionspecified by the user. Parameter (pept2protCombinationMethod) specifies whether the mean or the median of the quantification values is used to calculate the fold change between the two lists of peptides. End-users cannot change the default which is to use the mean. - Calculate the DEA significance indicator for each protein:
- yes (both the p-value and fold change are significant)
- p (only the p-value is significant)
- fc (only the fold change is significant)
- no (neither the p-value nor the fold change are significant)
alpha(significant means p \<alpha) andFCThreshold(significant means log2(condition/reference) >FCThreshold), respectively. - Save all results to disk and pass them on to the Report & visualization procedure.
DE test: moderated t-test
We would like to perform a moderated t-test (as used in limma for RNA-seq) for each protein between the quantification values of the reference condition and each other condition to see whether their means differ. In order to do this, for each protein we first average its associated peptide quantifications within the same sample because they are repeated measurement, and then for each condition pool the values of all associated samples, across all MS runs. This does mean we are not using all available information, which impacts statistical power, but exploiting that information requires a mixed model approach. We explicitly do not want to employ an approach using linear mixed models, as it is a cumbersome and time-consuming procedure for a data set with a few thousand proteins.
To control the false discovery rate – and not the family-wise error rate, as the Bonferroni correction does – we employ the Benjamini-Hochberg correction and obtain adjusted p-values.
When calculating the mean quantification value per condition for a certain
protein
over all its measurements, we have to take the nanmean to ignore missing
values. In
the case that there is just one value available for both conditions under consideration, the
result of the (moderated)
t-test is nonsensical and this protein is removed from the data frame after all.
Quality Control
- Combine only the quantification and Sequence columns of the data frames of all MS runs into one data frame, but this time as an 'inner join' on the peptide sequence. This way, only peptides observed in at least one sample of each MS run are present. They occur only once, and they only have missing values in quantification columns (which are all still distinguishable) if they were not detected in the corresponding sample. The result is a data frame with each unique peptide sequence that was found at least one in each condition across all MS runs.
- Perform a Principal Component Analysis (PCA) and Hierarchical Clustering (HC) on the transpose of the quantification matrix associated with the data frame from the previous step. Missing values will be imputed to be 0, because PCA and HC cannot handle them and zero- imputation does make sense in this situation. After all, the missing values that now remain are not due to a peptide not being present in a certain MS run or condition (because such entries have been removed during the construction of this data frame). That means they are missing quite probably because they are actually zero, or just very small (so that they did not meet the detection threshold). Therefore we deem this zero-imputation justified.
- Save all results to disk and pass them on to the Report & visualization procedure.
Principal component analysis
Performing a principal component analysis involves calculating the single-value
decom-
position (SVD) of the transposed quantification matrix across all the MS runs, which
is of size Pc × sum of all Ni , where Pc is
the amount of peptides found at least once in all
MS runs and conditions, and Ni is the amount of samples in MS run i. The
standard matplotlib function is rather slow for a matrix of this size, and
therefore we
will use the custom SVD method described by Halko et al. (DOI: 10.1137/090771806) and borrow
an
open
source implementation from GitHub.
Only the first two PCs are extracted.
Hierarchical clustering
A hierarchical clustering is performed on the transposed quantification matrix across all MS runs, so that each sample represents an observation and each peptide quantification is an observable (just like with the PCA). As a linkage criterium UPGMA (mean linkage) was used, together with a Euclidian metric.
The Report & visualization step takes the protein-level data frames with differential expression results, the PCA and HC results as well as other QC information and plots, the meta-data and application log file as input for the following steps.
- For each DE test, calculate a list of significantly (according to adjusted p-value) differentially expressed proteins, sorted according to adjusted p-value. For each protein, also provide the description, log2 fold change, adjusted p-value and total amount (before averaging repeated measurements) of supporting peptides observed. If both minimally and fully expressed protein data frames are given, create two lists of mutually exclusive proteins and add it to the meta-data for a consecutive sensitivity analysis.
- For each DE test, make a volcano plot of the minimal and/or full data frame,
with coloring
accord-
ing to significance level (see last step of DE), and
with the
protein IDs indicated
for all data points of the significance levels specified by
labelVolcanoPlotAreas. End-users cannot change this behavior and by default only data points with a 'yes' significance level are labeled. - Make a PCA plot, which is a 2D scatterplots of the first (horizontal axis) and second (vertical axis) principal component scores, where data points are colored according to condition and with different marker symbols for each MS run. The units on the axes bear no meaning other than dimensionless similarity measures.
- Make a HC Dendrogram plot, which is a dendrogram displaying the results of the hierarchical clustering, where singletons are colored according condition. The length of the dendrogram lines indicate the Euclidean distance between two leaves, although they bear no meaning other than a dimensionless similarity measure.
- Make an MS1 calibration plot, which is a scatterplot of the PSMs' relative (w.r.t. the maximum) PSM Engine Scores versus their "DeltaM [ppm]" value. Information from each tandem-MS run is plotted in a different color.
- Make an MS1 intensity histogram, which shows the amount of detected PSMs in function of their MS1 intensity. A histogram including information from only those PSMs actually used by IsoQuaC (i.e. not discarded for some reason).
- Generate a HTML report using flask, which contains the volcano plot(s), PCA plot, HC plot, list of top n differential proteins (sorted on adjusted p-value and with n specified by numDifferentials) and meta-data, possibly with additional info from the log file.
- Generate a PDF report from the HTML file and create a .zip-file with the DEA data frames in .tsv-format for each non-reference condition.
- Return all output files from the previous step to the web interface, which will display them on the front-end as well as e-mail them.
The PCA plot and HCD serve as quality control tools, as they give
the user an immediate impression of the influence of a sample belonging to a certain
condition as opposed to belonging to a certain MS run. If the normalization was
successful and there are sufficient differences between the conditions, one ought to
see
samples grouped together per condition (and not per MS run or some other criterium).
Also, when the samples corresponding to a certain condition are spread across multiple
MS runs, one could be worried that one particular sample is – for
whatever reason, biological or accidental or statistical – not like all the other samples
that belong to the same condition, which would distort the true statistics. However if such an
anomaly were to occur, it would be visible from the PCA and/or HC plot.
Technical Specifications
Input file formats
The workflow requires the following input files. The ones to be uploaded by the user are marked with (U), the others are automatically generated when using the web interface.
- (U) a Design of Experiments (DoE) file (can also be created using the web interface);
- (U) one PSM quantification data file per MS run (PD produces the right format);
- (U) (optional) a wrapper file when using custom data headers.
- one processing configuration file per MS run with the IsoQuaC processing settings;
- a job configuration file, auto-generated by the web interface based a.o. on some user-specified settings;
- (currently not available; optional) a file with the isotope impurities in matrix or TMT table format.
Design of Experiments file
This file is supposed to be uploaded by the user, who may create one from scratch or by using the DoE creator. The schema of the experimental setup is a tab-separated text file (.tsv or tabular .txt) with a layout like the example shown in the table below. For each separate tandem-MS run a name is be specified which will function as a prefix if no quanColumn aliases are provided (see further).
| MS run name | Condition:quanColumns:aliases | Condition:quanColumns:aliases | ... |
|---|---|---|---|
| TMT8_tuesday | mouse:126,127:m1,m2 | human:128,129:h1,h2 | ... |
| iTRAQ8_2 | human:114,115:h3,h4 | mouse:116,117: | ... |
| ... | ... | ... | ... |
For each MS run one can specify an arbitrary number of conditions, which do
not have to be in the
same column of the DoE file in case the same condition appears in multiple MS runs.
Each condition entry - which corresponds to one field in the file / table - is formatted as
follows:
first the name of the condition, followed by a colon, followed by a comma-separated list of
all
data frame quantification column names, optionally followed by a colon, optionaly followed
by a
comma-separated list of aliases for those quantification column names (in the same order).
These quantification column names and corresponding aliases will be automatically appended
to the
wrapper file, so there is no need to add them manually.
If no aliases are specified, the aliases are automatically generated to be the original
names
prefixed by the MS run's name and an underscore symbol. For instance, for the second
condition
of the iTRAQ8_2 MS run in the example table, the generated aliases would be
'iTRAQ8_2_mouse_116' and
'iTRAQ8_2_mouse_117'.
PSM files (quantification data)
The PSM files (maximum combined size: 75MB, can be changed on request) may be formatted as .xlsx (Microsoft Office), .csv (comma-separated), .tsv (tab-separated) or .txt (custom delimiter) files. By default, Proteome Discoverer (PD) produces tab- separated .txt files, and its PSM output files have names suffixed with “PSMs” should meet all requirements specified above. If you are using a different platform, read this section carefully and also take a look at the quick fixes in case you are unsure about how to proceed.
The following columns/quantities are absolutely required in each data file and should not contain missing values (the corresponding detections/rows will be discarded).
| Column name | Type | Details |
|---|---|---|
| Sequence | string | The raw peptide sequence as a sequence of 1-letter amino-acid representations, without modifications or any other annotations. |
| Master Protein Accessions | string; string; ... | UNIPROT identifiers of the proteins associated with a PSM, separated by '; ' (semi-colon and space). The master protein is the representative protein of choice for a whole protein group to which a peptide may correspond, as provided by PD2.1. |
| First Scan | integer | Identifier for the MS scan in which the peptide was detected. If you don't have such a column, just create one with all values set to a unique integer. |
| <quantification columns> |
float | Column names: channel names you provided in the
DoE
file. Values: Quantification values (strictly positive; zeros are replaced by NaN). We recommend using non-transformed intensities. Do not use log2(ratio) values. |
The above list also includes the names of the reporter quantification samples present in your data, although only one of them is required to contain an actual non-missing value in order not to be discarded. One can use either PD's intensities or S/N values as quantification values, since IsoQuaC can handle both and we find they produce practically indistinguishable results.
The following columns/quantities are only required under the circumstances mentioned and should in that case not contain missing values (the corresponding detections/rows will be discarded):
| Column name | Type | Details |
|---|---|---|
| Identifying Node Type | Mascot; Sequest HT; ... | (algorithm used to generate the PSM) if you used multiple PSM scoring algorithms. |
| Charge | integer | (charge of the precursor ion) if you wish to avoid aggregation on Charge (see Processing aggregation step) or do not care about missing values. |
| Modifications | string: mod1; mod2; ... | (Chemical modifications present on the peptide sequence) if you wish to avoid aggregation on Modifications (see Processing aggregation step) or do not care about missing values. The format should be a single string, suing ';' as a separator between multiple modifications. Information outside parentheses () is excluded automatically (e.g. PTM location information in the PD format). |
| Protein Accessions | string | (Uniprot identifiers of all matching protein according to the ‘Identifying Node’) if you wish to have a 'descriptions' column in your DEA output with Master Protein Descriptions; |
| Protein Descriptions | string | (Uniprot description of each matching protein) if you wish to have a 'descriptions' column in your DEA output with Master Protein Descriptions; |
| XCorr/Ions Score/... | float | (SEQUEST/MASCOT/... PSM score) if using
‘bestMatch’ as
the aggregate_method value (see Processing
aggregation step) and used SEQUEST/MASCOT/... to calculate PSMs, or if
you wish to get MS1 calibration QC plot; |
| Confidence | string: Low, Medium, High | (confidence level of the PSM identification) if
you wish
(removeBadConfidence_bool) to filter out PSMs with low confidence
values (removeBadConfidence_minimum); |
| Isolation Interference [%] | float | (measure for the relative amount of
co-isolation) if you
wish (removeIsolationInterference_bool) to filter out PSMs with
high isolation interference values
(removeIsolationInterference_threshold). |
| DeltaM [ppm] | float | (MS1 mass error) if you wish to get QC information w.r.t. the error on the precursor ion mass. |
| Ion Inject Time [ms] | float | (necessary MS2 ion accumulation time) if you wish to get QC information w.r.t. the necessary MS2 ion accumulation time. |
Other platforms
At this moment IsoQuaC is not really vendor-neutral, but we aspire to make it so. In the meanwhile, the following non-exhaustive list of suggestions may provide users with quick fixes so they can use the workflow even with different platforms:
- use a wrapper file to automatically transform column headers and increase your ease-of-use;
- if your PSMs do not have information about the first scan number, fake this column and fill it with unique integers;
- if your PSMs do not have information about charge or modifications, fake these columns and fill them with zeroes or empty strings, respectively; Caution: do not fill them with "None" or "N/A" strings or equivalent, as they will then be interpreted as missing values.
- if your PSM algorithm does not provide PSM scores, fake a score column filled with zeroes;
- if your PSM algorithm provides positive scores where ‘lower’ means ‘better’, negate all scores.
Wrapper file
The optional wrapper file is simply a .tsv- or tab-separated .txt-file with two
columns that
map original variable
names (first column) onto new ones (second column). Each line thus contains both a variable
name
from the user's custom data file and a new name – which ought to be one from the list
specified
by the wantedColumns parameter (see next section) – separated by a tab
character.
The quantification column names and corresponding aliases from the DoE file will
be automatically extracted and appended to the wrapper, so there is no need to add them
manually.
Processing Configuration file
Each processing configuration file contains the settings to be used in the
corresponding
Processing step and is generated automatically when using the web interface.
Such files are .ini files which contains only a [DEFAULT] section
and which is read using a ConfigParser from the configparser module, with options
allow_no_values=True, comment_prefixes=’;’ and
inline_comment_prefixes=’@’.
All possible parameters are listed in the table below, although none of these
settings can be explicitly changed by the end-user.
| Parameter name | default; other values | Details |
|---|---|---|
data |
Path on disk to the quantification data file. Auto-generated by the web interface from the DoE file. | |
delim_in |
\t | When uploading .txt files, it is advisable to explicitly specify the delimiter used, although the workflow does a pretty good job at auto-detecting it. |
wrapper |
Path on disk to the wrapper file. Auto-generated by the web interface from the DoE file. | |
quanColumns |
Columns headers of the quantification values. Auto-generated by the web interface from the experimental setup schema. | |
wantedColumns |
["Confidence", "Identifying Node Type", "Sequence", "Modifications", "Master Protein Accessions", "Protein Accessions", "# Protein Groups", "Protein Descriptions", "m/z [Da]", "Charge", "Deltam/z [Da]", "Isolation Interference [%%]", "RT [min]", "First Scan"]; [custom] | List of columns that are extracted from the PSM
data
files. All other columns are removed. This step happens after the
wrapper has been applied. The quanColumns are automatically
appended by the web interface. |
requiredColumns |
["First Scan", "Sequence", "Master Protein Accessions"]; [custom] | List of columns which are absolutely required
for the
workflow to complete successfully. The quanColumns are
automatically appended by the web interface. |
noMissingValuesColumns |
["First Scan", "Sequence", "Identifying Node Type", "Master Protein Accessions", "Charge", "Modifications"]; [custom] | List of columns in which no missing values may
appear.
If a missing value is found, the corresponding row is removed. The
quanColumns are automatically appended by the web interface.
|
removalColumnsToSave |
["First Scan", "Sequence", "Identifying Node Type", "Master Protein Accessions"]; [custom] | List of columns to save to disk whenever data is
removed
from the data frame for any reason (except during Aggregation). The quanColumns are
automatically appended by the web interface. This information will only ever be
used (anonymously) for meta-analysis purposes. |
aggregateColumnsToSave |
["First Scan", "Sequence", "Identifying Node Type", "RT [min]", "Charge", "Modifications", "Master Protein Accessions", "Degeneracy"]; [custom] | List of columns to save to disk whenever data is
removed
from the data frame during Aggregation.
The
quanColumns are automatically appended by the web interface. This
information will only ever be used (anonymously) for meta-analysis purposes.
|
removeBadConfidence_bool |
true; false | Remove PSMs with ‘Confidence’ lower than
removeBadConfidence_minimum.
|
removeBadConfidence_minimum |
Medium; High; Low | PSMs with ‘Confidence’ lower than this value are
removed
if so specified by removeBadConfidence_bool |
removeIsolationInterference_bool
|
true | Remove PSMs with ‘Isolation interference [%]’
lower than
removeIsolationInterference_threshold.
|
removeIsolationInterference_threshold
|
30; float between 0 and 100 | 2 PSMs with ‘Isolation interference [%]’ higher than this value are removed if so specified by removeIsolationInterference_bool |
PSMEnginePriority |
String-encoded Python dictionary of PSM detection algorithms and their score column names used while removing redundancy due to the use of multiple PSM algorithms/engines. They are ordered according to priority (lower list index means higher priority). | |
PSMEnginePriority: engineNames |
[unspecified]; [Mascot, Sequest HT]], [custom, custom, ...], ... | Names of PSM algorithms/engines, ordered
according to
priority (lower list index means higher priority). If set to
"unspecified", the workflow assumes there is no PSM Algorithm
information available and removePSMEngineRedundancy_bool has no
effect. |
PSMEnginePriority: scoreNames |
[unspecified]; [Ions Score, XCorr], [custom, custom, ...], ... | Score columns names corresponding to the list of PSM algorithms/engines, in the same order. |
removePSMEngineRedundancy_bool |
true; false | Remove redundancy due to the use of multiple PSM algorithms. Behavior of the workflow with value 'false' has not been tested. |
removePSMEngineRedundancy_exclusive_bool
|
false; true | Controls whether only PSMs detected by the master algorithm should be kept. |
(Currently disabled)
isotopicCorrection_bool
|
false | Perform isotopic correction using isotopicCorrection_matrix or not. |
(Currently disabled)
isotopicCorrection_matrix
|
Path to the isotope impurity matrix. Auto-generated by the web interface after uploading an isotope impurity file. | |
aggregate_method |
bestMatch; mostIntense, mean, geometricMedian | The method specifying how to combine quantification values of multiple representee PSMs. |
aggregateRT_bool |
true; false | Remove redundancy due to different retention times. |
aggregateCharge_bool |
true; false | Remove redundancy due to different charge states. |
aggregatePTM_bool |
false; true | Remove redundancy due to different post-translational modifications. |
precision |
10-5; float | Desirable precision of the CONSTANd algorithm. |
maxIterations |
50; integer > 0 | Maximum number of iterations of the CONSTANd algorithm. |
removedDataInOneFile_bool |
false; true | If enabled, saves all data frames with information about removed data in separate files. |
path_out |
Path on disk to save output files to. Auto-generated by the web interface from the experimental setup schema. | |
filename_out |
result | Auto-generated by the web interface from the experimental setup schema. |
delim_out |
\t | Text delimiter used when writing Processing output to disk. |
Job configuration file
The job configuration file contains the settings to be used in the corresponding
Analysis and Report steps and is generated automatically when using the web interface, in
which
case only the parameters marked with (U) can be adjusted by the user.
It is a .ini file which contains only a [DEFAULT] section
and which is read using a ConfigParser from the configparser module, with options
allow_no_values=True, comment_prefixes=’;’ and
inline_comment_prefixes=’@’.
All possible parameters are listed below, together with their {default values; other
possible
values} and descriptions.
| Parameter name | default; other values | Details |
|---|---|---|
| date | now | Full Python datetime (%Y-%m-%d %H:%M:%S.%f) at upload of DoE file, auto-generated by the web interface. |
| schema | String-encoded Python dictionary containing the DoE information as well as some additional meta-information. Auto-generated by the web interface from the experimental setup schema and uploaded files. | |
| getRTIsolationInfo_bool | false, true | If enabled, gathers RT isolation statistics during the RT Aggregation step and stores it in metadata. This information will only ever be used (anonymously) for meta-analysis purposes. |
| pept2protCombinationMethod | mean; median | Central measure to generate one quantification value for a protein from a list of peptide quantification values when calculating fold changes for the DEA. |
| minExpression_bool | true; false | Do a DEA for the minimally expressed set of proteins. |
| (U) fullExpression_bool | false; true | Do a DEA for the fully expressed set of proteins. |
| (U) referenceCondition | Name of the condition to be used as the reference in the DEA. | |
| alpha | 0.05; float between 0 and 1 | Significance level for the differential expression's moderated t-test. |
| FCThreshold | 1; float > 0 | Threshold above which a fold change value is considered significant in th DEA. |
| labelVolcanoPlotAreas | [true, false, false, false]; ... | Significance levels whose proteins to label with their protein IDs. Order: [‘yes’, ‘p’, ‘fc’, ‘no’]. |
| PCA_components | 2; integer ∈ {2..amount of conditions} | Number of principal components to be extracted in the PCA |
| (U) numDifferentials | 10; integer > 0 | Number of top differentially expressed proteins to show in the report. |
| (U) jobname | Name of the job, specified by the user. Will be appended to the timestamp to form the job ID. | |
| path_out | Path on disk to where output files ought to be saved. | |
| (U) delim_out | \t; any character | Text delimiter used when writing Analysis and Report output to disk. |
| jobID | date_jobname | Concatenation of job date and job name with underscore separator. Auto-generated by web interface. |
| (U) mailRecipient | E-mailadress to which output and job status updates are sent. |
Schema structure
The schema in the table above is a String-encoded, nested Python dictionary with the key-value structure shown below, where 'channel' is synonymous for 'quantification column'.
| Key | Value / Nested Key | ... | ... |
|---|---|---|---|
| allMSRuns | [MS runs] | ||
| allConditions | [conditions] | ||
| MSRun | allMSRunConditions | [conditions] | |
| allMSRunChannelNames | [channelNames] | ||
| allMSRunChannelAliases | [channelAliases] | ||
| config | path | ||
| data | path | ||
| wrapper | path | ||
| isotopicCorrection_matrix | (disabled) path | ||
| {conditions} | channelNames | [names] | |
| channelAliases | [aliases] | ||
| {...} | {...} | [...] |
(disabled) Isotope impurities file
For each data file one may optionally specify the isotope impurities using another .tsv- file. This file may either contain a matrix (used directly by the workflow) or a TMT-style formatted table (IDT) that is automatically transformed into the correct matrix by the web interface. In the former case this is a tab-separated isotope impurity matrix (IIM). In the latter case the format is also tabular, resembling the format used in the Thermo Fisher's COA documents and with the string “IDT” as the columns header for the sample names.
Contact us
Contact us through: [email protected]